
婴儿型黑矇性痴呆别名:晚发性婴儿黑矇性家族性痴呆
1.临床表现 两性均可罹患,起病于1~4岁,突然出现严重惊厥。常呈肌阵挛性或无动作发作。运动和智力发育落后、视力减退、肌张力减低、共济失调、视网膜萎缩、黄斑变性、视神经萎缩而渐致全盲。患儿之肌阵挛发作用抗惊厥药物往往无效。常有脑小畸形。出现手足徐动、瘫痪、握持反射和颈肢反射已属晚期。
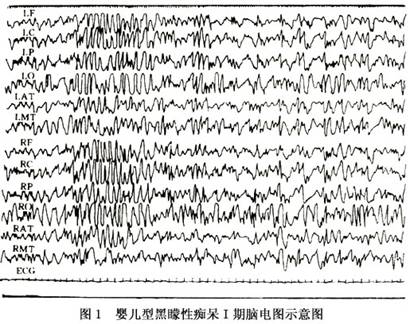
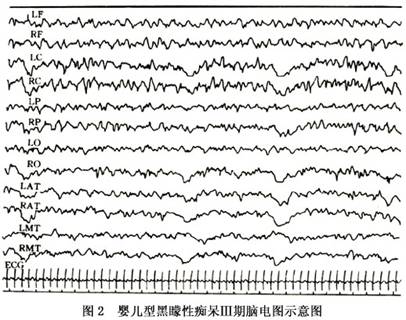
2.临床分期 Morell和Torrss(1960)将本病按临床经过分为4期,并对各期的临床表现及脑电图所见进行了探讨(表1)。

第Ⅱ期:1~1.5岁,肌张力开始增高,腱反射亢进,出现紧张性颈反射。有时伴全身强直性抽搐。脑电图以1~2Hz的高波幅慢波为主,有时频繁出现两侧性棘慢波综合,酷似高度失律。
第Ⅲ期:1岁4个月~2岁,经常出现肌阵挛发作,对周围事物漠不关心,视力明显减退乃至失明。脑电图示波幅降低,爆发局灶性癫痫波。无觉醒反应(图2)。
第Ⅳ期:2岁以后,完全失明,经常出现全身性抽搐。脑电图示低波幅活动,但癫痫波逐渐消失(图1,2)。
3.临床分型
(1)Tay-Sachs病(Ⅰ型GM2神经节苷脂沉积症):典型的临床表现是在4~6个月正常发育后,患儿出现精神运动发育迟缓及退化,对声音有惊跳反应、肌张力低、对周围环境兴奋降低,头不能自控,淡漠发生早、樱红斑可能较晚。惊厥发生较晚。在疾病的进展期,病人对外界刺激反应差,头大,在最后阶段头明显增大。无内脏增大发现。
本病多见于东欧犹太人,但在诊断时不能除外非犹太人及非白人儿童患病。
诊断依赖于氨基己糖酶同工酶A的测定。本症无特殊治疗,一般在3~4岁死亡。
(2)Sandhoff病(Ⅱ型GM2神经节苷脂沉积症):临床表现类似Tay-Sachs病,但有内脏受累。
(3)少年型GM2神经节苷脂沉积症(Ⅲ型):发病较Tay-Sachs病及Sandhoff病晚。共济失调和进行性精神运动发育迟缓始于2~6岁。语言丧失、进行性强直状态,手和四肢呈徐动姿势,并有细小痉挛发生,并未发现器官增大、骨骼畸形及空泡细胞,在晚期可失明。患儿多在5~15岁死亡。
根据临床表现特点和实验室检查确诊。
与GML神经节苷脂沉积症鉴别。
该症自婴儿期发病,是一种进行性脑变性疾病及黏多糖病样外形为其特征的疾病。
GML神经节苷脂是一种单唾液酸神经节苷脂,存在于正常大脑皮质和白质中,在内脏中也有少量,在多唾液酸神经节苷脂正常的分解代谢中也可形成。
由于β-半乳糖苷脂酶活性的缺乏,使GML神经节苷脂分解代谢发生障碍,致使GML神经节苷脂发生沉积。在脑中沉积可致严重的神经细胞损害,并伴脱髓鞘和神经胶质增生。受累的神经显示有膜状胞浆小体(cytoplasmic membranous bodies),就像在Tay-Sachs病所见到的。本症为常染色体隐性遗传。
推荐药店

婴儿型黑矇性痴呆找问答
暂无相关问答!
婴儿型黑矇性痴呆找药品
暂无相关药品!
用药指南
暂无相关用药指导!